Tutorial
A simple two atoms system of N2 is shown as example. With the following code a N2 molecule is first optimized, then a frequency analysis is performed and lastly, the molecule is stretched.
from ase import Atoms
from ase.calculators.emt import EMT
from ase.optimize import BFGS
from ase.vibrations import Vibrations
#create the structure
n2 = Atoms('N2', [(0, 0, 0), (0, 0, 1.1)])
#define the code/metod that is responsible for the electronic structure calculation
calc = EMT()
n2.set_calculator(calc)
#do a geometry optimization
BFGS(n2).run(fmax=0.01)
#do a frequency analysis
vib = Vibrations(n2)
vib.run()
hessian = vib.get_vibrations()
#distort the structure and get the energy
n2l = n2.copy()
n2l.positions[1][2] = n2.positions[1][2]+0.1
n2l.calc = EMT()
n2l.get_potential_energy()
from strainjedi.jedi import Jedi
j = Jedi(n2, n2l, hessian)
j.run()
This will give following output
Step Time Energy fmax
BFGS: 0 09:10:42 0.440344 3.251800
BFGS: 1 09:10:42 0.264361 0.347497
BFGS: 2 09:10:42 0.262860 0.080535
BFGS: 3 09:10:42 0.262777 0.001453
************************************************
* JEDI ANALYSIS *
* Judgement of Energy DIstribution *
************************************************
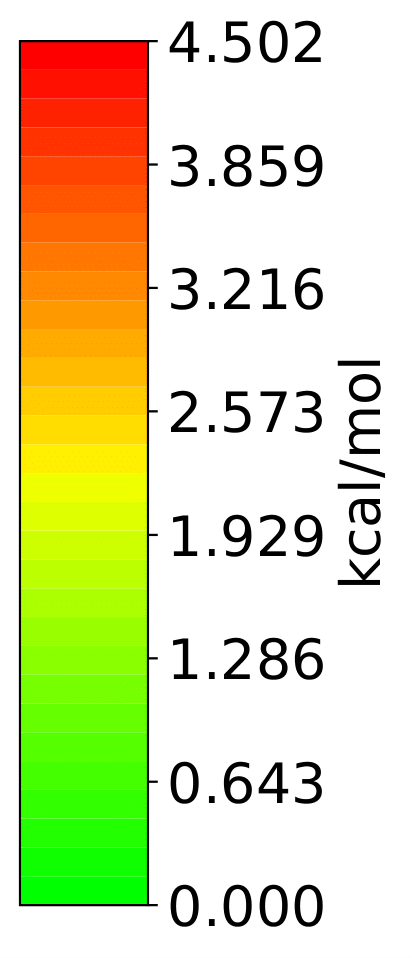
Strain Energy (kcal/mol) Deviation (%)
Geometries 3.95089665 -
Red. Int. Modes 4.50191012 13.95
RIM No. RIM type indices delta_q (au) Percentage Energy (kcal/mol)
1 bond N0 N1 0.1889726 100.0 4.5019101
RIM stands for redundant internal mode, delta_q is the strain in each RIM.
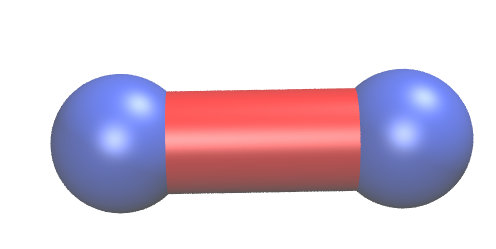
j.vmd_gen()
generates a vmd folder with files that are VMD scripts ‘bl.vmd’, ‘all.vmd’ (, ‘ba.vmd’, ‘da.vmd’), pictures of colorbars ‘blcolorbar.png’, ‘allcolorbar.png’, …, energies calculated for each bond E_bl, E_all, …, and the xyz geometry of the strained structure xF.txt.